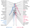We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
RNA category is based on mRNA expression levels in the analyzed samples (RNA assay description). The categories include: tissue/cell line enriched, group enriched, tissue/cell line enhanced, expressed in all, mixed and not detected. RNA category is calculated separately for The Cancer Genome Atlas (TCGA) data from cancer tissues and internally generated Human Protein Atlas (HPA) data from normal tissues and cell lines.
TCGA (cancer tissue):
Expressed in all
HPA (cell line):
Mixed
HPA (normal tissue):
Expressed in all
Protein evidencei
Protein evidence scores are generated from several independent sources and are classified as evidence at i) protein level, ii) transcript level, iii) no evidence, or iv) not available.
Evidence at protein level
Protein expression normal tissuei
A summary of the overall protein expression pattern across the analyzed normal tissues. The summary is based on knowledge-based annotation.
"Estimation of protein expression could not be performed. View primary data." is shown for genes analyzed with a knowledge-based approach where available RNA-seq and gene/protein characterization data has been evaluated as not sufficient in combination with immunohistochemistry data to yield a reliable estimation of the protein expression profile.
Standardized explanatory sentences with additional information required for full understanding of the knowledge-based expression profile.
Antibody staining mainly consistent with RNA expression data.
Reliability score - normal tissuesi
Reliability score (score description), divided into Enhanced, Supported, Approved, or Uncertain, is evaluated in normal tissues and based on consistency between antibody staining pattern, available RNA-Seq and gene/protein characterization data, as well as similarity between independent antibodies targeting the same protein.
Kaplan-Meier plots for all cancers where high expression of this gene has significant (p<0.001) association with patient survival are shown in this summary. Whether the prognosis is favourable or unfavourable is indicated in brackets. Each Kaplan-Meier plot is clickable and redirects to a detailed page that includes individual expression and survival data for patients with the selected cancer.
RNA expression overview shows RNA-seq data from The Cancer Genome Atlas (TCGA).
TCGA dataseti
RNA-seq data in 17 cancer types are reported as median FPKM (number Fragments Per Kilobase of exon per Million reads), generated by the The Cancer Genome Atlas (TCGA). RNA cancer tissue category is calculated based on mRNA expression levels across all 17 cancer tissues and include: cancer tissue enriched, cancer group enriched, cancer tissue enhanced, expressed in all, mixed and not detected. To access cancer specific RNA and prognostic data, click on the cancer name. The cancer types are color-coded according to which type of normal organ the cancer originates from.
Antibody staining in 20 different cancers is summarized by a selection of four standard cancer tissue samples representative of the overall staining pattern. From left: colorectal cancer, breast cancer, prostate cancer and lung cancer. An additional fifth image can be added as a complement. The assay and annotation is described here. Note that samples used for immunohistochemistry by the Human Protein Atlas do not correspond to samples in the TCGA dataset.
For each cancer, color-coded bars indicate the percentage of patients (maximum 12 patients) with high and medium protein expression level. The cancer types are color-coded according to which type of normal organ the cancer originates from. Low or not detected protein expression results in a white bar. Mouse-over function shows details about expression level and normal tissue of origin. The images and annotations can be accessed by clicking on the cancer name or protein expression bar. If more than one antibody is analyzed, the tabs at the top of the staining summary section can be used to toggle between the different antibodies.
Most thyroid cancers and a few hepatocellular carcinomas showed strong cytoplasmic positivity. Other malignancies showed weak to moderate cytoplasmic immunoreactivity.
GENE INFORMATIONi
Gene information from Ensembl and Entrez, as well as links to available gene identifiers are displayed here. Information was retrieved from Ensembl if not indicated otherwise.
Gene name
ALDH9A1 (HGNC Symbol)
Synonyms
ALDH4, ALDH7, ALDH9, E3
Description
Aldehyde dehydrogenase 9 family member A1 (HGNC Symbol)
Entrez gene summary
This protein belongs to the aldehyde dehydrogenase family of proteins. It has a high activity for oxidation of gamma-aminobutyraldehyde and other amino aldehydes. The enzyme catalyzes the dehydrogenation of gamma-aminobutyraldehyde to gamma-aminobutyric acid (GABA). This isozyme is a tetramer of identical 54-kD subunits. [provided by RefSeq, Jul 2008]
The protein browser displays the antigen location on the target protein(s) and the features of the target protein. The tabs at the top of the protein view section can be used to switch between the different splice variants to which an antigen has been mapped.
At the top of the view, the position of the antigen (identified by the corresponding HPA identifier) is shown as a green bar. A yellow triangle on the bar indicates a <100% sequence identity to the protein target.
Under the antigens, the maximum percent sequence identity of the protein to all other proteins from other human genes is displayed, using a sliding window of 10 aa residues (HsID 10) or 50 aa residues (HsID 50). The region with the lowest possible identity is always selected for antigen design, with a maximum identity of 60% allowed for designing a single-target antigen (read more).
The curve in blue displays the predicted antigenicity i.e. the tendency for different regions of the protein to generate an immune response, with peak regions being predicted to be more antigenic.The curve shows average values based on a sliding window approach using an in-house propensity scale. (read more).
If a signal peptide is predicted by a majority of the signal peptide predictors SPOCTOPUS, SignalP 4.0, and Phobius (turquoise) and/or transmembrane regions (orange) are predicted by MDM, these are displayed.
Low complexity regions are shown in yellow and InterPro regions in green. Common (purple) and unique (grey) regions between different splice variants of the gene are also displayed (read more), and at the bottom of the protein view is the protein scale.
ALDH9A1-001
PROTEIN INFORMATIONi
The protein information section displays alternative protein-coding transcripts (splice variants) encoded by this gene according to the Ensembl database.
The ENSP identifier links to the Ensembl website protein summary, while the ENST identifier links to the Ensembl website transcript summary for the selected splice variant. The data in the UniProt column can be expanded to show links to all matching UniProt identifiers for this protein.
The protein classes assigned to this protein are shown if expanding the data in the protein class column. Parent protein classes are in bold font and subclasses are listed under the parent class.
The Gene Ontology terms assigned to this protein are listed if expanding the Gene ontology column. The length of the protein (amino acid residues according to Ensembl), molecular mass (kDalton), predicted signal peptide (according to a majority of the signal peptide predictors SPOCTOPUS, SignalP 4.0, and Phobius) and the number of predicted transmembrane region(s) (according to MDM) are also reported.
 The Human Protein Atlas project is funded
The Human Protein Atlas project is funded



 MENU
MENU