We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
The protein expression bar, with the units not detected (n), low (l), medium (m) and high (h), is based on a best estimate of the true protein expression for proteins where the antibody staining pattern has been analyzed by knowledge-based annotation. For genes where more than one antibody has been used, a collective score is set. For details, see Assays & annotation.
h
m
l
n
RNA expressioni
RNA expression shows average values based on RNA-seq data generated by HPA, the Genotype-Tissue Expression (GTEx) consortium or the FANTOM5 consortium.
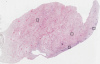
The detailed normal tissue page shows images of the stained tissue, together with antibody staining and expression level of the cell types. A knowledge-based annotated protein expression is provided for each cell type at the top of the page and the staining level of the individual antibodies is given underneath each antibody ID.
Samples from up to three different individuals have been stained for each antibody. The gender, age and tissue characterization are reported for each individual (patient) and are viewable when clicking on the image for magnification.
The images can be clicked for an enlarged view that can be panned. From the enlarged view, all stained images for all antibodies can be browsed (represented by miniature images). The miniature image with an orange overlay is the currently displayed image.
This score describes the level of antibody staining observed in the annotated cell types as not detected, low, medium, or high. It is based on the staining intensity and fraction of stained cells.
The RNA-seq details section shows detailed information about the individual samples used for the transcript profiling and results of the RNA-seq analysis.
Information about each individual sample is listed below, including gender, age, a tissue section image and estimated fractions of cell types. TPM (transcripts per million) values give a quantification of the gene abundance which is comparable between different genes and samples.
RNA-Seq data is reported as average RPKM (reads per kilobase per million mapped reads), generated by the Genotype-Tissue Expression (GTEx) project. More information can be found on the GTEx portal.
Normal distribution across the dataset is visualized with box plots, shown as median and 25th and 75th percentiles. Points are displayed as outliers if they are above or below 1.5 times the interquartile range. RPKM values of the individual samples are presented next to the box plot.
Max RPKM: 2.8
Min RPKM: 1.8
Std RPKM: 0.3
Median RPKM: 2.6
GTEX-S32W-1526-SM-4AD6Z
50-59 years, female
2.8
GTEX-U3ZN-1626-SM-4DXTZ
30-39 years, female
2.8
GTEX-T5JW-0726-SM-4DM6D
20-29 years, female
2.7
GTEX-S341-1126-SM-4AD6T
40-49 years, female
2.5
GTEX-S4UY-1426-SM-4AD6Y
40-49 years, female
2.4
GTEX-TSE9-2826-SM-4DXTF
60-69 years, female
1.8
Cervix - Endocervix RPKM: 2.9
Samples: 5
Max RPKM: 4.0
Min RPKM: 2.0
Std RPKM: 0.7
Median RPKM: 3.0
GTEX-S341-1326-SM-4AD72
40-49 years, female
4.0
GTEX-S32W-1626-SM-4AD6G
50-59 years, female
3.1
GTEX-T6MO-1426-SM-4DM73
40-49 years, female
3.0
GTEX-TML8-0726-SM-4DXTT
40-49 years, female
2.2
GTEX-TSE9-2726-SM-4DXSQ
60-69 years, female
2.0
CERVIX, UTERINE - FANTOM5 CAGEi
Tissue data obtained through Cap Analysis of Gene Expression (CAGE) are reported as Tags Per Million, generated by the FANTOM5 project. More information can be found here.
 The Human Protein Atlas project is funded
The Human Protein Atlas project is funded



 MENU
MENU